

10 Last-Minute Questions for QMSR Compliance
We're less than a month away from the QMSR. Are you ready?


Only paid subscribers get regular full access to our guidance breakdowns and other analyses. If you’re not already a paid subscriber, you can upgrade here.
By now, most FDA-regulated medical device firms understand the headline:
The Quality Management System Regulation (QMSR) is replacing the legacy Quality System Regulation (QSR) at the beginning of February.
What’s less clear (especially this late in the transition) is whether firms can demonstrate compliance in a way that will hold up during an FDA inspection.
At this stage, having helped many firms access the SMEs they need to make that transition, success is not about whether you’ve “mapped to ISO 13485.” It’s about whether your quality system is coherent, documented, implemented, and defensible under FDA scrutiny (without long explanations, interpretive bridges, or “we’re still transitioning” caveats).
Below, we’ve laid out 10 questions every impacted firm should be asking itself right now, along with prescriptive guidance on what the FDA will likely expect to see when they walk in.
(Also, be aware that the FDA recently published a new final rule updating references across 21 CFR Parts 801, 803, 812, 860, 862, 864, 866, 868, 872, 874, 876, 878, 880, 882, 886, 888, 890, and 892 to align the broader medical device regulatory framework with the previously finalized QMSR. Read our other post on that here.)
If you still need to get compliant with the QMSR, talk to us ASAP. We’ll pair you with the ISO/FDA expertise that can be hard to find for projects like this.
1. Is your quality management system explicitly documented as ISO 13485-compliant under QMSR?
QMSR §820.10 requires manufacturers to document a QMS that complies with ISO 13485 as incorporated by reference — not merely operate one in practice.
The FDA emphasized that ISO 13485 now forms the foundational CGMP framework, but compliance with ISO alone is not sufficient unless the FDA’s supplemental provisions are also met. This includes clearly documenting:
The scope of your QMS.
Your regulatory responsibilities.
Any justified exclusions or non-applications.
How risk-based controls are applied across processes.
If your quality manual still reads like a lightly updated Part 820 document, this is an immediate red flag! We’ve been helping firms go back in and make the more substantive documented changes that regulators are communicating their expectations around.
We recommend updating your quality manual to explicitly state ISO 13485:2016 compliance and documenting your regulatory role(s) under applicable FDA requirements.
Identify and justify any exclusions or non-applications, and also make sure the quality manual describes QMS process interactions, not just document lists.
2. Have you embedded risk-based thinking across the QMS (or confined it to design controls)?
One of the biggest shifts reflected throughout the move from the QSR to the QMSR is the pervasive role of risk under ISO 13485. Risk is referenced repeatedly across management responsibility, design controls, purchasing, training, software validation, CAPA, and supplier oversight. Having seen inside the systems of many device firms, we know firsthand that most firms will need to bolster risk management across their quality system.
The FDA will not accept risk language that’s generic, cosmetic, or isolated to design controls only.
You should be able to demonstrate how risk actually informs your decisions, including:
Process controls
Supplier evaluation
Training effectiveness
Software validation scope
CAPA prioritization
If risk does not visibly drive how you’re allocating resources in these areas, the FDA will see the gap immediately. This is one area where we spend the most time in our QMSR remediation projects with clients. Risk has to stretch into more systems than what’s been acceptable until now.
A few specific action items here if you know there’s work to do:
Map where risk influences decisions across the QMS.
Ensure supplier qualification, audit frequency, and training depth are risk-proportionate.
Document risk considerations in CAPA prioritization and investigation depth.
Confirm risk management activities are recorded and retrievable.
3. Does top management actively demonstrate QMS ownership beyond policy approval?
ISO 13485 strengthens the expectations around management commitment, and the QMSR will reinforce this through FDA enforcement. Investigators will expect to see evidence that executive leadership:
Communicates regulatory and quality importance internally.
Sets measurable quality objectives.
Reviews QMS performance with defined inputs and outputs.
Acts on identified improvement needs.
Management review under ISO 13485 is far more structured than the legacy QSR. It’s one of the areas where there’s quite a bit of difference between old and new. If your reviews are informal, irregular, or poorly documented, this is a common inspection finding waiting to happen.
Make sure you’re:
Defining measurable quality objectives tied to regulatory performance.
Documenting how leadership communicates regulatory importance internally.
Making sure your management review inputs and outputs meet ISO 13485 specificity.
Recording decisions, actions, and follow-up (not just meeting minutes).
4. Do you have a compliant quality manual, and does it actually reflect how the QMS operates?
A formal quality manual is now a requirement. Under ISO 13485 and QMSR, the quality manual must describe:
The structure of the QMS.
Documented procedures or references to them.
Interaction between QMS processes.
The scope of the system and justifications for exclusionsThe .
The FDA will likely compare your quality manual to what they observe in practice. Discrepancies between documented intent and operational reality often trigger deeper inspection scrutiny.
If you haven’t already:
Confirm your quality manual includes scope, exclusions, procedures, and process interactions.
Cross-check the manual against your actual operational practices.
Eliminate any “aspirational” language that is not supported by records. (This is surprisingly common in quality manuals we look at.)
Prepare the quality manual as an inspection navigation tool. Could you use it to guide the inspection you're hosting right now? This is a sign of a strong manual.
5. Are internal audits risk-based, planned, and followed through?
ISO 13485 steps up internal audit expectations, and QMSR brings those requirements squarely into the FDA’s enforcement scope now. The agency will expect to see a planned and documented audit program that sets an audit frequency based on risk and scope.
They’ll also want to see fully-documented corrective actions and timely follow-up and verification of effectiveness of those corrective actions.
In our opinion, for the vast majority of device firms, a single annual audit covering everything equally is unlikely to pass scrutiny under a risk-based framework.
As part of our QMSR compliance work, we’ve been helping firms set up that risk-based audit schedule, document the audit scope rationale, track corrective actions through verification, and ensure processes are in place so management reviews audit outcomes, not just summaries. Talk to us if there’s work to do here.
6. Can you prove personnel competence (not just training completion)?
Training under ISO 13485 goes beyond attendance. It requires demonstrated competence, proportionate to risk. You should be able to show:
How training needs are actually determined in the first place.
How effectiveness is evaluated.
How risk influences the depth of the training you’re doing.
How competency is maintained over time.
Firms often underestimate how frequently the FDA challenges training effectiveness during inspections. Now the expectations will be even higher.
Make sure you define the competence criteria for quality-impacting roles and are evaluating training effectiveness, not just attendance. Now is a good time to check on how well you’re keeping those training records.
7. Are design controls aligned to ISO 13485 without losing FDA-critical DHF expectations?
While ISO 13485 uses different terminology (e.g., “design and development files”), the FDA still expects clear design history documentation, traceable approvals, reviews, verification, and validation—and risk management outputs integrated into design inputs and changes.
The QMSR allows flexibility in how design reviews are structured, but documentation rigor remains non-negotiable.
A few important points here for compliance:
Make sure you’re maintaining DHF-equivalent documentation, even if it’s renamed now.
Integrate risk management outputs into design inputs and changes.
Make sure all verification and validation records are complete and traceable.
Avoid assuming ISO terminology eliminates FDA documentation expectations!
8. Do supplier controls reflect risk, services, and outsourced processes, not just materials?
ISO 13485 expands purchasing controls to include services, software, outsourced processes, and quality agreements where applicable. Under QMSR, the FDA will expect supplier evaluation and re-evaluation to be:
Risk-based
Ongoing
Documented
Linked to purchasing controls and CAPA when needed
This is a common gap area we see for firms that rely heavily on consultants, CROs, CMOs, or software vendors.
Be sure you’re classifying your suppliers by risk—including service providers! Also, document supplier evaluation and re-evaluation criteria and make sure you’re linking any supplier issues to actual CAPA processes.
9. Are complaints, servicing, and postmarket records FDA-ready, not just ISO-compliant?
Several of the QMSR supplemental provisions reinforce FDA-specific recordkeeping requirements, including:
Complaint files
MDR decision logic
Service records
Accessibility of records for FDA inspection
U.S. availability of records for foreign manufacturers
ISO 13485 alone does not fully satisfy these expectations. Firms must explicitly address the FDA’s supplemental record controls.
Verify that your complaint files include all FDA-required data elements and make sure MDR decision logic is documented. Also:
Confirm that service records meet the QMSR minimum content requirements.
Validate U.S. accessibility of records for any foreign manufacturers.
10. Could you defend our QMS (end-to-end) in a real FDA inspection soon?
The most important question is not whether your system maps to ISO 13485 on paper. It’s whether:
Records are complete, current, and retrievable.
Risk decisions are defensible.
Management oversight is visible.
Gaps are known and actively addressed.
FDA investigators will not conduct a theoretical standards comparison. While we’re still waiting to see what the FDA will replace the QSIT with, it’s reasonable to assume they’ll test how your QMS performs under pressure.
Once you wrap your transition, be sure to pressure-test record retrieval and traceability and make sure leadership can articulate how the QMS works in practice.
Download our QMSR gap analysis worksheet
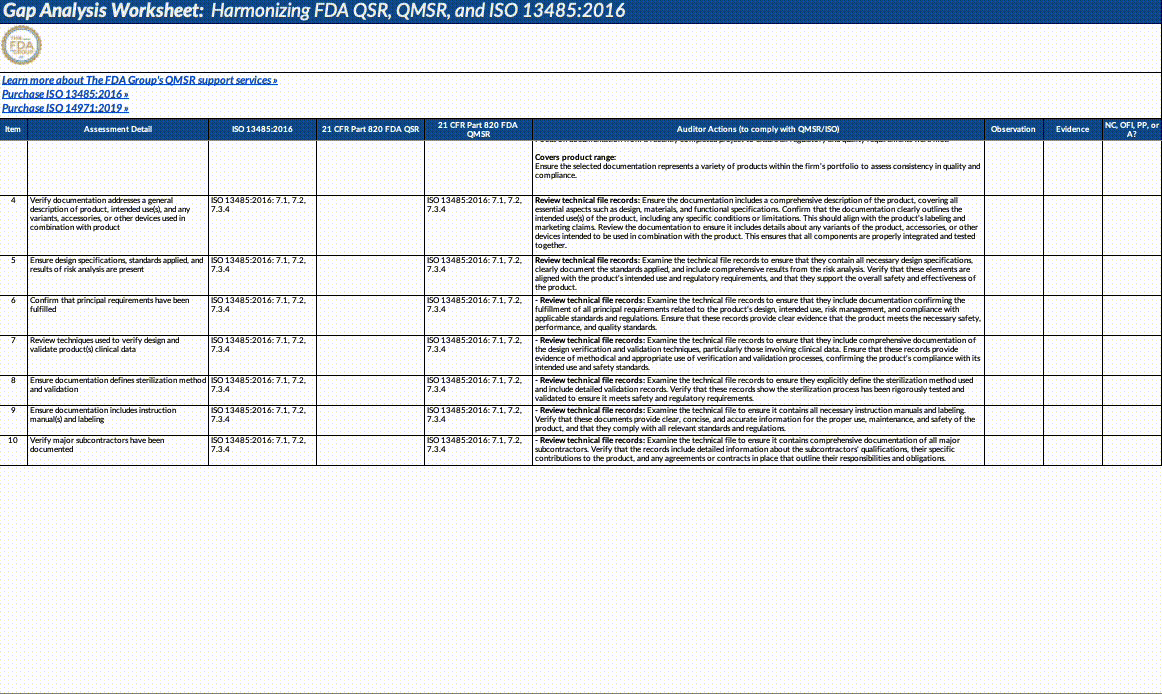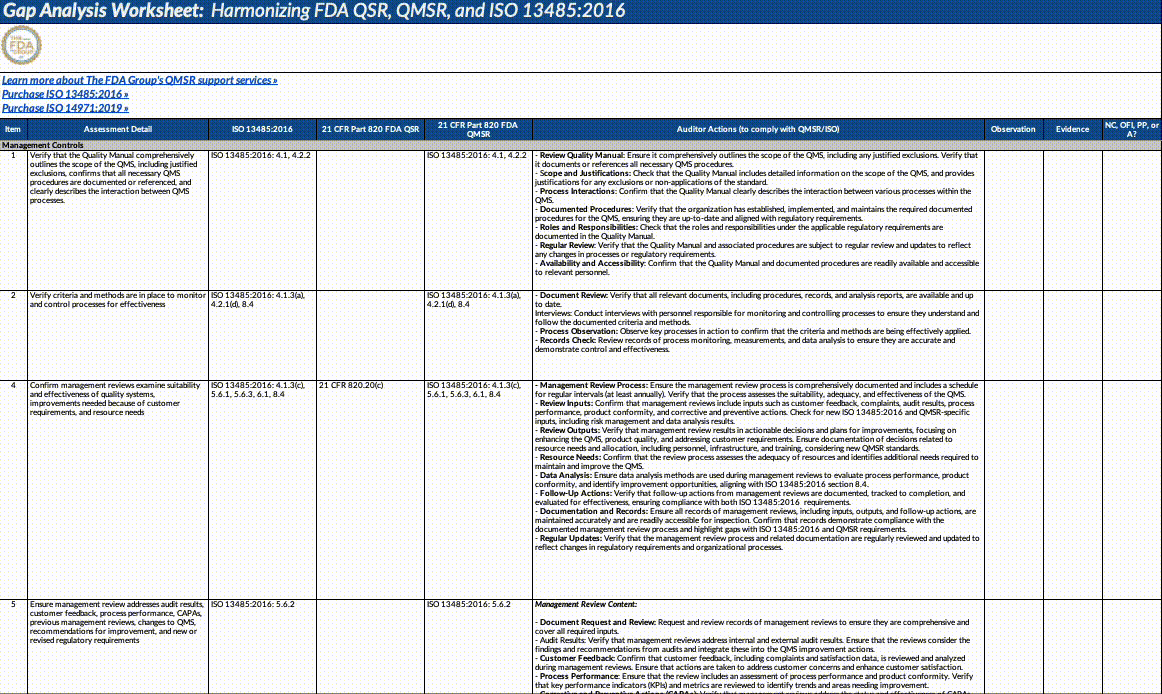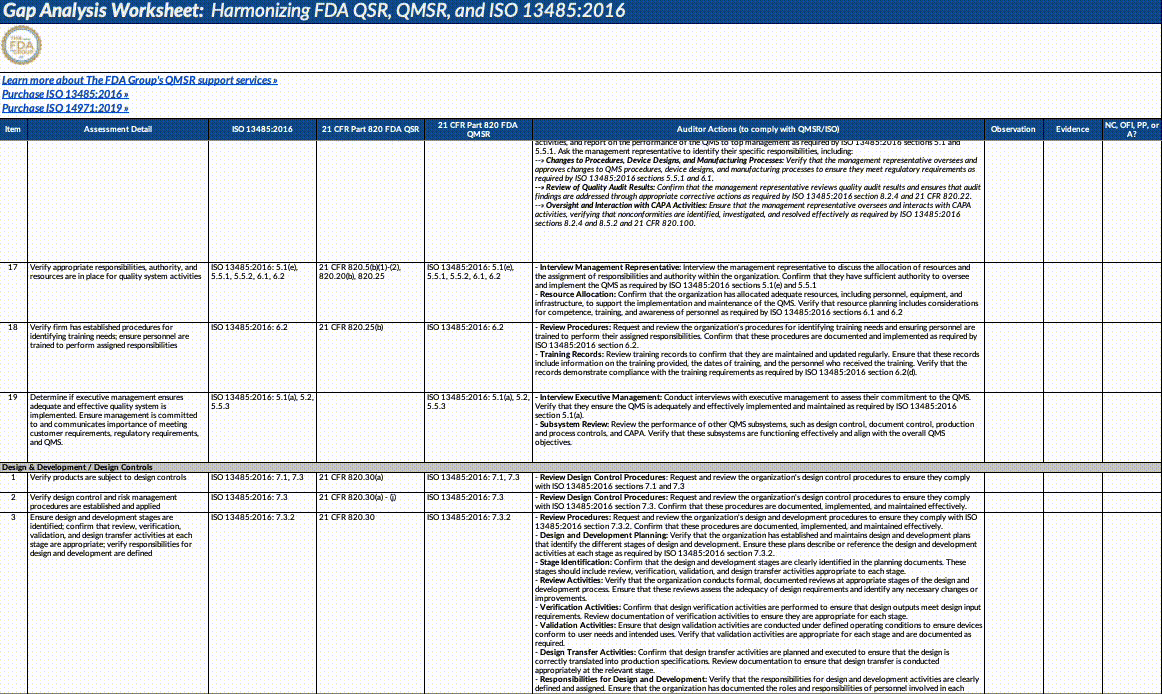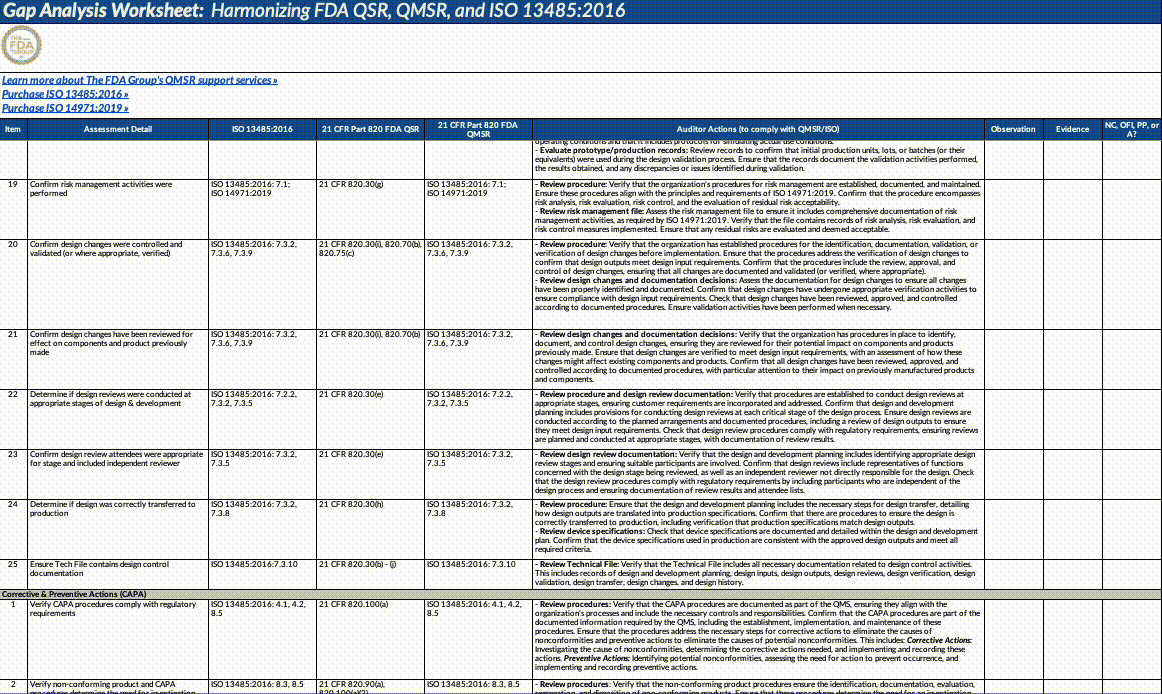
If you’re a paid subscriber, grab our Excel worksheet below to run your own QMSR gap analysis line-by-line. (Free subscribers will see a paywall. Upgrade here.)
