

10 Takeaways from FDA's QMSR Town Hall on Risk-Based Inspections
A quick breakdown of FDA's April town hall on risk-based medical device inspections under the new Compliance Program 7382.850.


This one’s for our device teams out there. By now, you should definitely know that the FDA is now actively inspecting to the QMSR, which went live in February.
At the beginning of April, the Agency held its latest town hall on the regulation, which focused on how inspections will actually work under the updated Inspection of Medical Device Manufacturers Compliance Program Manual (CP 7382.850).
If you missed it, this is the session you can’t afford to skip! The FDA brought a panel of compliance and inspectorate leaders who answered questions that have been circulating across the industry since the transition.
The panel included:
Karen Masley-Joseph (Senior Advisor, Office of Medical Device and Radiological Health Inspectorate)
Keisha Thomas (Associate Director for Compliance & Quality, CDRH)
Tonya Wilbon (Assistant Director for Postmarket Industry Education, DICE)
CAPT Kimberly Lewandowski-Walker (Regulatory Officer, FDA Inspections and Regulatory Audits Team)
Here’s everything they said — distilled, organized, and translated into action items your team can use right now.
1. The QSIT is gone, and the new inspection framework is live
This is no longer a transition. As of February 2, 2026, the FDA has fully retired the Quality System Inspection Technique (QSIT) and the two prior compliance programs that governed device inspections (CP 7382.845 and CP 7383.001). Everything — surveillance inspections, PMA preapproval and postmarket inspections, compliance follow-ups, for-cause inspections — now runs through a single document: Compliance Program 7382.850, Inspection of Medical Device Manufacturers.
If you haven’t downloaded and read this document yet, stop here and do that first.
It’s publicly available as a PDF on the FDA’s CDRH Compliance Programs webpage. As Karen Masley-Joseph emphasized during the presentation, this isn’t guidance; it’s the FDA’s internal procedure for how investigators conduct inspections. But the Agency publishes it for transparency, and there is no reason not to use it to prepare.
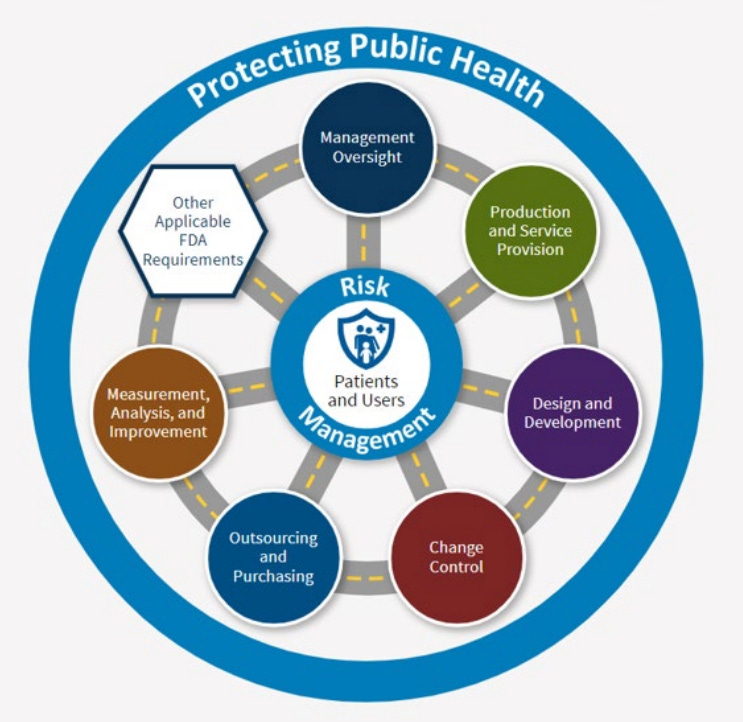
2. Every inspection now covers your entire QMS
“One difference between this inspection process and QSIT is that now, on every inspection, FDA will evaluate, to some extent, requirements in each of the main parts of your quality management system.” — Karen Masley-Joseph
Under QSIT, inspections focused on specific subsystems. An investigator might concentrate heavily on CAPA or production controls and never touch other areas. That’s over, according to the FDA.
Under CP 7382.850, every inspection (whether it’s a routine surveillance visit or a compliance follow-up) will evaluate requirements across all six QMS Areas and the four Other Applicable FDA Requirements (OAFRs).
The six QMS Areas are:
Management Oversight
Design and Development
Production and Service Provision
Change Control
Outsourcing and Purchasing
Measurement, Analysis, and Improvement
The four OAFRs are:
Medical Device Reporting
Reports of Corrections and Removals
Medical Device Tracking Requirements
Unique Device Identification
How deep investigators go in each area depends on which inspection model applies. But the days of having entire sections of your QMS go unreviewed during a given inspection seem to be over.
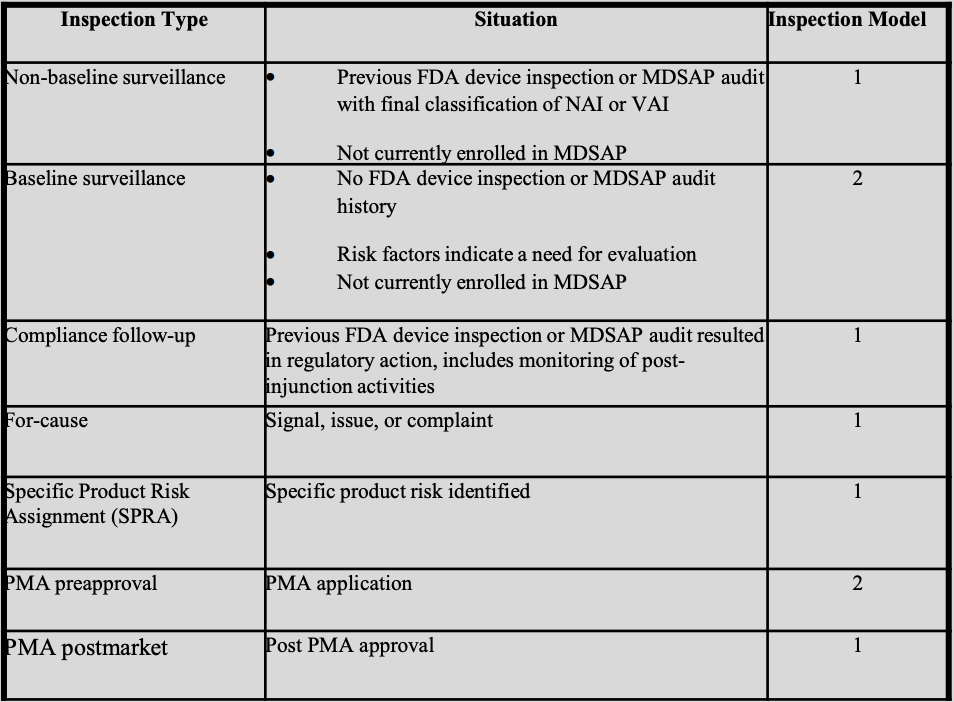
3. There are two actual inspection models
The FDA has organized its new device inspections into seven types, each mapped to one of two inspection models.
Model 1 (most common)
Model 1 applies to non-baseline surveillance, compliance follow-up, for-cause surveillance, specific product risk assignment (SPRA), and PMA postmarket inspections.
Under Model 1, investigators must evaluate at least one element in each of the six QMS Areas and the applicable OAFRs. This is the minimum.
The FDA was explicit during the panel discussion that investigators will typically cover more than the minimum and that the approach is intentionally flexible.
CAPT Lewandowski-Walker stressed two words manufacturers should internalize: “minimum” and “flexible.” She noted that going beyond the minimum number of elements should not be thought of as expanding the scope of the inspection. It’s how the risk-based approach is intended to work.
Model 2 (more comprehensive)
Model 2 applies to: baseline surveillance and PMA preapproval inspections.
Under Model 2, investigators must evaluate specific named elements within each QMS Area, not just “at least one.”
This is a significantly more comprehensive review. For example, under Design and Development alone, Model 2 requires evaluation of inputs, outputs, review, verification, validation, software validation, and transfer. All applicable OAFRs are also reviewed, unless it’s a PMA preapproval where the device isn’t yet on the market.
4. Risk management is now central to every part of the inspection
“While investigators previously have requested risk management documentation, under the QMSR they will emphasize reviewing the risk management documents and evaluating how manufacturers have effectively implemented risk controls throughout their quality management system.” — Karen Masley-Joseph
This was probably the loudest message of the entire town hall. Under the former QSR, investigators requested risk management documentation, but it wasn’t the through-line of the inspection. Under QMSR, it is!
The FDA’s stated inspection goal now has two parts:
Evaluate whether the manufacturer’s QMS meets FDA requirements and provides reasonable assurance of device safety and effectiveness.
Evaluate whether risk management and risk-based decision making are effectively used in the QMS.
That second piece is new emphasis. During the inspection, investigators will review risk management files and reports, risk analyses identifying hazards and harms, documentation showing how risk controls were implemented, and evidence that those controls are actually effective. They’ll trace risk documentation throughout the product lifecycle.
To be extra clear here, the FDA will emphasize risk management documents and risk controls throughout the QMS, not just in the design file.
5. No need for separate risk assessments of administrative processes
This was one of the most practical clarifications from the panel, and it’s one that should relieve a lot of anxiety.
You do not need separate risk assessments for administrative processes like document control or training.
Karen Masley-Joseph addressed this directly:
"Manufacturers do not need to create separate risk assessments or risk management documents for these, like administrative processes, like document control or training."
The risk-based approach required under Clause 4.1.2(b) does apply to all QMS processes, including administrative ones. But this is different from the formal risk management processes required under Clause 7.1 for product realization. Manufacturers don’t need to create standalone risk management documents for things like training frequency or document control. Instead, when making decisions about these processes, they should reference existing product-related risk management documentation and document those risk-based decisions.
The example FDA gave: if you’re deciding how often to retrain staff on a manufacturing procedure, you don’t need a formal risk assessment for “training frequency.” Instead, reference your existing risk documentation for that manufacturing process. If the process is high risk, that should inform a decision to train more frequently or more rigorously.
The FDA’s inspection focus will be on whether manufacturers are using product-related risk information to make appropriate decisions, not on whether you’ve created a separate risk assessment for every administrative function.
6. No more statistical sampling — investigators will review records as they see fit
Under QSIT, investigators followed a defined statistical sampling plan when reviewing records. That requirement is now gone.
Tonya Wilbon confirmed that investigators will now conduct records review — not statistical sampling — and will review records as they deem appropriate. The number and type of records reviewed will be based on identified product risk and the investigator’s professional experience and judgment. If an investigator’s findings during the inspection lead them to concerns, they may review additional records.
"The simple answer is actually no... the investigators will conduct records review, and not just not sampling of records. So they will review records as they deem appropriate during the inspections." — Tonya Wilbon
This is a meaningful change. It gives investigators broader discretion, and it means manufacturers can’t rely on the probability that a specific record won’t be selected in a sample. Your documentation needs to be consistently solid.
7. Management review, internal audits, and supplier audits are now fair game
Under the old QSR, records related to management review, internal audits, and supplier audits were exempt from routine inspection review under 21 CFR 820.180(c). That exemption no longer exists under the QMSR.
"The exemption that previously existed for these records is not a part of QMSR... But as noted, FDA does not expect manufacturers to revise or recreate records made before February 2nd, 2026 to comply with QMSR requirements. But they are still fair game for us to look at if the inspection direction leads us there." — Keisha Thomas
For baseline surveillance and PMA preapproval inspections, these records will be specifically requested. For other inspection types, investigators will use their findings to determine when to review them. She gave a practical example: if an inspection reveals design deficiencies that resulted in a recall, investigators may assess how the manufacturer has been auditing its design and development process.
The FDA also confirmed that investigators may review records created before February 2, 2026, including management review and internal audit records. However — and this is important — the FDA does not expect manufacturers to revise or recreate pre-QMSR records to comply with the new requirements. You don’t need to go back and add ISO 13485 references to old documents or remove QSR-era terminology like “design history file” or “device master record.”
If a review of your pre-QMSR records reveals gaps in your QMS (such as failure to incorporate feedback into risk management) those gaps need to be addressed now.
8. Notified Body audits can complement, but can’t replace, internal audits
The FDA was clear:
"A Notified Body audit that focuses on ISO 13485 conformity assessment may not replace an internal audit of the QMSR requirements as the QMSR requirements include all the requirements for 21 CFR 820." — Tonya Wilbon
ISO 13485 certification from a Notified Body does not equal compliance with the QMSR. And Notified Body audits cannot replace your internal audit program.