

QSIT is Officially Replaced: What FDA’s New Inspection Program Means Under QMSR
Effective today, FDA medical device inspections will no longer be conducted under the Quality System Inspection Technique (QSIT).


It’s QMSR day for those in the device and medtech space. And it comes with some big news on the inspection side: The FDA has now formally replaced the Quality System Inspection Technique (QSIT) with a new Compliance Program Manual for Inspection of Medical Device Manufacturers (CP 7382.850), effective February 2, 2026, alongside implementation of the QMSR.
The new compliance program formalizes how FDA investigators are instructed to plan, scope, execute, and escalate inspections under QMSR. For manufacturers, it provides the clearest picture yet of how FDA intends to evaluate quality systems in practice once QMSR is fully in force.
Below, we break down at a high level what’s changed, what hasn’t, and where we expect inspections to become more rigorous.
Talk to us if you need audit, mock inspection, or general inspection-readiness support to align with these new expectations.
Why the FDA replaced the QSIT
To start with some background, the QSIT was built around a subsystem-based inspection model that reflected the structure of the Quality System Regulation (QSR). While it allowed flexibility, it was never fully aligned with:
Risk-based inspection mandates under FDARA.
The lifecycle thinking emphasized in recent legislation.
The FDA’s increasing reliance on benefit-risk principles.
ISO 13485–style process integration.
The new compliance program explicitly states that inspections must now:
Encompass the total product lifecycle (TPLC).
Be risk-based, with inspection depth driven by patient and user risk.
Evaluate whether risk management and risk-based decision making are actually functioning inside the QMS, not merely documented.
Risk now starts with the patient
One of the most important additions to our eye in the new manual is a formal inspection model diagram that places patients and users at the center of FDA medical device inspections.
From there, FDA investigators are instructed to use a manufacturer’s risk management documentation to decide:
Which QMS areas to prioritize.
Which records to sample.
How deeply to evaluate controls.
Inspections are explicitly tied to product risks that could adversely impact patients or users, and inspectors are expected to connect findings across processes, not evaluate requirements in isolation.
In practical terms, this means the FDA is no longer “checking systems.” It’s following risk signals through your quality system.
From QSIT subsystems to QMS areas
Under QSIT, inspections revolved around subsystems like CAPA, Production & Process Controls, and Design Controls. Under the new compliance program, inspections are organized around six QMS Areas plus four Other Applicable FDA Requirements (OAFRs).
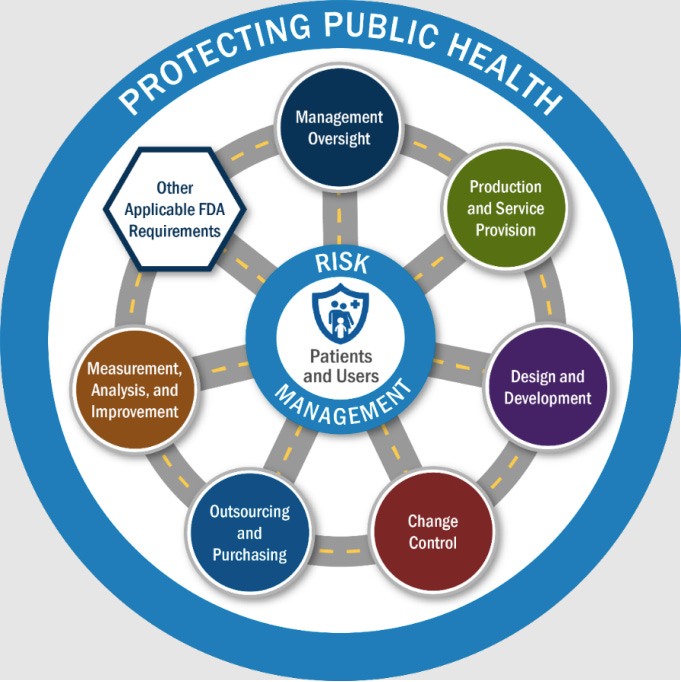
The six QMS areas
FDA inspections must now evaluate requirements across:
Management Oversight
Design and Development
Production and Service Provision
Outsourcing and Purchasing
Change Control
Measurement, Analysis, and Improvement
Each QMS Area contains multiple elements, and each element maps to one or more regulatory requirements. Importantly, inspectors are not required to evaluate these areas in a fixed order. They are instructed to move between them as risk dictates.
Management oversight is no longer indirect
One of the most consequential changes in the new inspection program is how explicitly management oversight is brought into scope. The manual makes clear that the FDA may now review:
Management review records
Internal audit results
Supplier audit outcomes
Risk-based decision-making at the leadership level
This aligns with QMSR’s emphasis on top management responsibility and the FDA’s stated expectation that executive leadership actively ensures regulatory compliance through an integrated QMS.
For many organizations, this will be a shift. Historically, management review and internal audits were often treated as formalities—checked for existence but rarely scrutinized in depth. Under the new inspection model, these activities are central to the FDA’s assessment of whether a firm’s quality system is effective.
What has not changed
Despite the new structure, manufacturers should not expect inspections to become “lighter.” The manual is explicit that the FDA will continue to examine familiar areas, including:
Complaints and complaint handling
Medical Device Reporting (MDRs)
Corrections and removals
Recalls
Unique Device Identification (UDI)
Servicing activities
Design controls
Production processes
Supplier controls
The difference is how these areas are evaluated.
Instead of being reviewed as standalone subsystems, they are now assessed in context—connected through risk management, postmarket signals, and management oversight.
Risk-based scheduling and inspection depth
The new compliance program also formalizes how inspections are prioritized and scoped. The FDA will schedule inspections using a risk-based methodology that considers, among other factors:
PMA preapproval and postmarket status
Compliance history
Product risk classification
Frequency of recalls or MDRs
Newly marketed devices
Signals from postmarket data
Once onsite, inspectors are instructed to identify product risks before and during the inspection, select records and processes based on those risks, and then expand the inspection scope if objectionable conditions are found.
This creates a feedback loop where weak risk management can directly lead to broader inspection coverage.
Expect deeper scrutiny of internal controls
The manual also reflects the FDA’s expanded authority under recent legislation to request records in advance of or in lieu of inspections, and to evaluate internal systems more comprehensively.
Practically speaking, this means that poorly integrated QMS processes are more likely to be uncovered. Disconnects between risk management, CAPA, complaints, and management review may be easier to identify and superficial compliance activities are more likely to result in observations.
In our experience, firms that struggle under this more modern model are not necessarily those with the most deficiencies, but those whose systems do not tell a coherent risk story.
What manufacturers should be doing now
With QMSR and the new inspection program now fully defined, manufacturers should be asking:
Can we clearly explain how product risk drives our QMS activities?
Do management review and internal audits actually influence decisions?
Are supplier controls integrated into risk management (not siloed)?
Can we trace postmarket signals through CAPA, change control, and leadership oversight?
If the answer to any of these questions is unclear, inspections under CP 7382.850 are likely to feel more invasive than QSIT ever did.
If you need help preparing for QMSR-aligned inspections, pressure-testing management oversight, or responding to FDA observations under the new framework, this is exactly the work we support every day. Talk to us.
Who is The FDA Group?
The FDA Group helps life science organizations rapidly access the industry's best consultants, contractors, and candidates. Our resources assist in every stage of the product lifecycle, from clinical development to commercialization, with a focus on Quality Assurance, Regulatory Affairs, and Clinical Operations.
With thousands of resources worldwide, hundreds of whom are former FDA, we meet your precise resourcing needs through a fast, convenient talent selection process supported by a Total Quality Guarantee. Learn more and schedule a call with us to see if we’re a fit to help you access specialized professionals and execute your projects on time and on budget.