

Guidance Breakdown: Responding to FDA Form 483 Observations at the Conclusion of a Drug CGMP Inspection
What drug manufacturers need to know about the FDA's dedicated (draft) guidance on responding to Form 483 observations.


This guidance breakdown is available in full to paid subscribers. Only paid subscribers get regular full access to our guidance breakdowns and other analyses. If you’re not already a paid subscriber, you can upgrade here.
The FDA has released a draft guidance that every drug manufacturer responsible for inspection readiness should read closely: Responding to FDA Form 483 Observations at the Conclusion of a Drug CGMP Inspection.
This is the first time the FDA has issued a standalone guidance dedicated entirely to how establishments should respond to a Form 483.
For decades, expectations around Form 483 responses have largely been transmitted through experience rather than formal guidance: the accumulated practices learned by regulatory consultants, quality leaders, and former investigators through years of interacting with FDA field investigators, compliance officers, and district offices.
Having been on the frontline of this for years, we know some companies do it well. Some very well. Others really don’t. And a lot of what passes for a “483 response” would be better described as a loosely organized collection of promises. The FDA is now putting its expectations in writing, and in doing so, it’s codifying a level of rigor that many establishments are not currently meeting.
This guidance was jointly prepared by CDER’s Office of Compliance, CBER, CVM, and the Office of Inspections and Investigations (OII). It applies broadly: foreign and domestic drug manufacturing establishments, biologics manufacturers, veterinary drug facilities, 503B outsourcing facilities, and combination product manufacturers where CDER or CBER is the lead center.
Let’s break down what the FDA actually said, what they’re signaling, and where companies routinely get this wrong.
If you ever need expert 483 response or remediation support, don’t hesitate to contact us.
Quick context: what a 483 actually is (and isn’t)
Before we get into the response framework, it’s worth restating something the guidance itself makes clear: an FDA 483 contains inspectional observations that, in the investigator’s judgment, may constitute violations of the FD&C Act. It does not represent the FDA’s final findings or conclusions about an establishment’s compliance status.
This distinction matters because it shapes how a company should think about its response. The 483 is not a verdict! It’s the opening of a conversation — one that can end with no further action, or one that can escalate to a warning letter, import alert, consent decree, or worse. Your response is the single most influential variable in that trajectory. It’s why firms hire us to ensure it’s as strong as possible.
The FDA explicitly says that a written response may be the primary or key component in their review when deciding whether to pursue subsequent action. That’s about as close as the FDA gets to saying: this is your best shot to shape the outcome.
The 15-business-day window is tight
The guidance recommends that firms submit their response within 15 business days after the 483 is issued. This isn’t new, of course. The 15-day convention has been understood in the industry for years. But the guidance sharpens it in a way that should concern any company without a pre-built response infrastructure.
First, the FDA says that if a response arrives within 15 business days, it plans to conduct a detailed review before determining whether to pursue subsequent action. The flip side of that statement is equally important: the FDA will not ordinarily delay regulatory action, such as issuing a warning letter, to review a response received after 15 business days. In practical terms, missing that window means the Agency may proceed toward regulatory action without delaying its decision to conduct a detailed pre-action review of your response.
Second, and in some ways the operationally important, the FDA recommends addressing all observations in a single response rather than submitting piecemeal replies to individual observations. This runs counter to what some companies do: firing off partial responses as individual CAPAs are completed, thinking it demonstrates urgency. The FDA recommends submitting a single comprehensive response addressing all observations within the 15-business-day window rather than multiple piecemeal submissions.
For complex observations that can’t be fully addressed in 15 business days, the guidance recommends submitting a CAPA plan and a proposed timeframe for substantive responses within that 15-business-day window. This is critical! The FDA isn’t expecting every root cause to be identified and every corrective action completed in three weeks. They’re expecting you to (1) demonstrate that you understand the problem, (2) that you have a credible plan, and (3) that you can tell them when the rest is coming.
What the FDA actually wants in a 483 response
The response format section of this guidance is remarkably specific, and it should effectively serve as a template for any quality team building (or rebuilding) their 483 response process now and definitely once the guidance is finalized.
The FDA expects, at a minimum, the following:
Establishment identification. Name, full site address, and the FDA Establishment Identifier (FEI) number from the top of the 483. Basic, but it gets missed! Especially when responses are drafted by outside counsel or consultants who aren’t working from the original 483.
A copy of the 483 itself. Attached to the response. Again, simple, but it anchors the response to the specific observations.
Identity of the preparer and signatory. The FDA wants to know who wrote the response and, importantly, who signed it. The response should be signed by someone in executive management with the authority to allocate resources and implement commitments. Other key personnel, such as the site head or head of the quality unit, may also sign the response. Someone who can actually move money, hire people, and authorize capital projects. The guidance also allows the site head or head of quality to co-sign, but the executive accountability piece is clearly intentional.
Letters of authorization if the response was prepared by a consultant or outside counsel.
Global investigation plans and reports. If the investigation extends beyond the inspected site (and it often should), those broader plans need to be included.
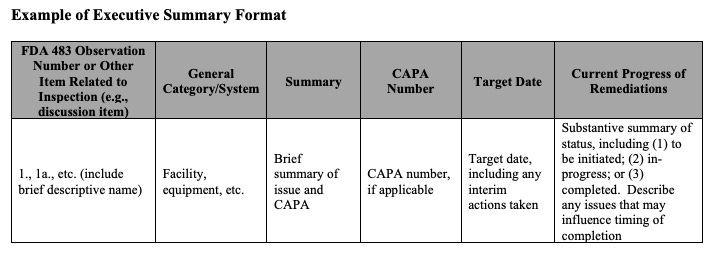
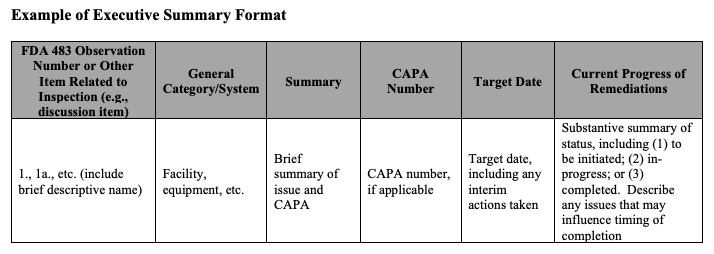
An executive summary with a specific structure. This is where the guidance gets most prescriptive. The FDA provides an example table format showing how the executive summary can be organized: observation number, general category or system, brief summary, CAPA number, target date, and current progress of remediation. The progress column should include whether each item is to be initiated, in progress, or completed, along with any issues that may influence timing.
Page 5 Detailed discussion of each observation. Each observation (or group of related observations) should have its own section, individually noted and numbered in the table of contents.
Within each, the FDA expects a patient- and product-focused risk assessment covering inventory and distributed drugs still within expiry, with an assessment of possible effects on safety, identity, strength, quality, and purity.
They also expect a detailed investigation report including scope, summary, associated drugs and lot numbers, identified root causes and systemic issues, the CAPA plan, completed actions and interim measures, and a planned effectiveness evaluation.
Signed attachments. Attachments should include signatures indicating support for the contents. Consultant-provided attachments should be signed by the consultant.
There’s also a quiet but important expectation embedded throughout: the FDA wants to see that you’ve gone beyond the specific observation and assessed whether the underlying issue affects other drugs, processes, facilities, or contract organizations. The guidance returns to this point repeatedly.
Going beyond the observation: our notes from the field
Let’s linger on this point a bit since it’s so critical.
In our experience, this expectation that you look beyond the four corners of the cited observation is one of the biggest differentiators between a 483 response that satisfies the FDA and one that invites a warning letter. We’ve supported manufacturers through enough post-inspection responses to know that this is where most companies underestimate what’s required.
A few examples of what adequate scope expansion looks like in practice (this is us talking now, not the guidance):
If the observation cites a failure in cleaning validation for one production line, the investigation shouldn’t stop at that line. You should evaluate whether the same cleaning procedures, SOPs, or validation protocols apply to other lines, other equipment trains, or other products manufactured on shared equipment. If they do (and they often do!), the scope of the investigation should include all of them, along with an assessment of whether the product already distributed may have been affected.
If the observation involves a data integrity finding in the QC lab (say, unauthorized deletions in a chromatography data system) the investigation needs to assess every system where the same access controls (or lack thereof) exist. That means audit trails across LIMS, ERP, and any electronic batch record systems, not just the single instrument the investigator happened to look at. The FDA’s Data Integrity Q&A guidance already points in this direction, and this new 483 response guidance reinforces it.
