

What Our Auditors Are Finding Lately: 10 Trends Across GMP, GCP, ISO, and GDP Audits (H2 2025)
A look back at our recent audit reports, mock inspections, gap assessments, and supplier qualifications.


A quick heads up before you dive in: This is a long, in-depth post. Some email providers (including Gmail) may clip it in your inbox. If that happens, just click “View entire message” or “Read on Substack” to see the full article.
About once a quarter (and more often if possible), we’ll be publishing an analysis of the trends we’re seeing in our audit work for clients, so (if you’re a paid subscriber) you can learn and act proactively on what we’re finding across the industry. In this issue, we’re looking at the whole second half of 2025.
If you’re a free subscriber, we’ll be cutting off the findings list and a list of 10 questions every firm should be able to answer with documentary evidence at hand. If the answer is “not right now,” you may have a vulnerability worth closing before your next inspection. Graduate to a paid subscription here.
Over the second half of 2025, our auditors conducted a diverse portfolio of engagements spanning pharma manufacturing, clinical supply chains, medical device operations, biologics production, and lab services across three continents.
These were deep dives into quality systems at companies ranging from emerging biotechs preparing for their first FDA pre-approval inspection to established global CDMOs handling hundreds of client audits per year.
What struck us wasn’t the critical findings (there were virtually none). Rather, it was the consistency of the minor and major observations. The same themes appeared again and again: documentation gaps that made otherwise solid processes look questionable, timeline management that seemed reasonable in practice but failed on paper, and procedural ambiguities that left companies exposed to regulatory risk they hadn’t fully recognized.
This report synthesizes those findings into practical guidance. We’ve anonymized every example to protect our clients, but we’ve preserved enough operational context that you should be able to see yourself—or your suppliers, or your CMOs—in these scenarios.
Talk to us if you need auditing, mock inspection, remediation, or other RA/QA/Clinical support.
What’s in this dataset
This trends report uses only the audit reports in our project set from July to November 2025. The set includes audit engagements across a few distinct contexts:
Mock Pre-Approval Inspections (PAI): Five-day mock PAIs at U.S.-based pharma sites preparing for ANDA submission, including DPI and SMI manufacturing.
Routine GMP Vendor Audits (API and Finished Dosage): Audits of high-volume generic manufacturers in India producing oral solids for the U.S. market under USFDA and MHRA approvals.
Supplier Qualification Audits: First-time and re-qualification audits at CDMOs in China providing API synthesis, drug product manufacturing, and ADC conjugation.
GLP-to-GMP Gap Assessments: Gap assessments at R&D laboratories transitioning to GMP-compliant batch release testing, evaluated against 21 CFR Part 211 and ICH Q7 to support Phase 3 and commercial readiness.
cGMP Packaging and Labeling Audits: Audits of clinical and commercial packaging and labeling operations supporting multinational trials.
Medical Device and Primary Packaging Audits: Audits of ISO 13485-certified device and packaging component manufacturers, including combination products and primary packaging, assessed against ISO 13485, ISO 15378, and 21 CFR Part 820.
Clinical Trial Sponsor Oversight (BIMO): Mock BIMO inspections for IDE studies evaluating sponsor oversight, monitoring, data integrity, and device accountability under 21 CFR Parts 812 and 820.
Secondary Packaging and Distribution Audits: Monitoring audits of logistics providers handling temperature-controlled storage, packaging, and global distribution under EU GDP and 21 CFR Part 211, with emphasis on thermal mapping and deviation management.
Again, no company or site names are used in the trend descriptions below, and we haven’t added any facts beyond what those reports contain. Confidentiality is paramount to us.
Stats and trends at a glance
The dataset:
33 audits analyzed across August–November 2025
5 countries: United States, India, China, Ireland, Netherlands
Audit types: GMP vendor qualification, mock PAI, gap assessments, ISO 13485 medical device, clinical packaging/GDP, mock BIMO, biologics/ADC CDMO
Regulatory frameworks: 21 CFR 210/211, 21 CFR 820, 21 CFR 58, ISO 13485, ISO 15378, EU Annex 13, ICH Q7/Q9/Q10
Finding severity breakdown:
Critical findings: 0 (0%)
Major findings: ~12 (~15%)
Minor findings: ~50 (~60%)
Recommendations: ~25 (~25%)
Top finding categories (by % of audits affected):
Documentation and change control — 70%
Supplier/vendor oversight — 45%
Investigation and CAPA timeliness — 40%
Facility and equipment gaps — 35%
Complaint handling backlogs — 25%
Data integrity vulnerabilities — 25%
APR/management review gaps — 20%
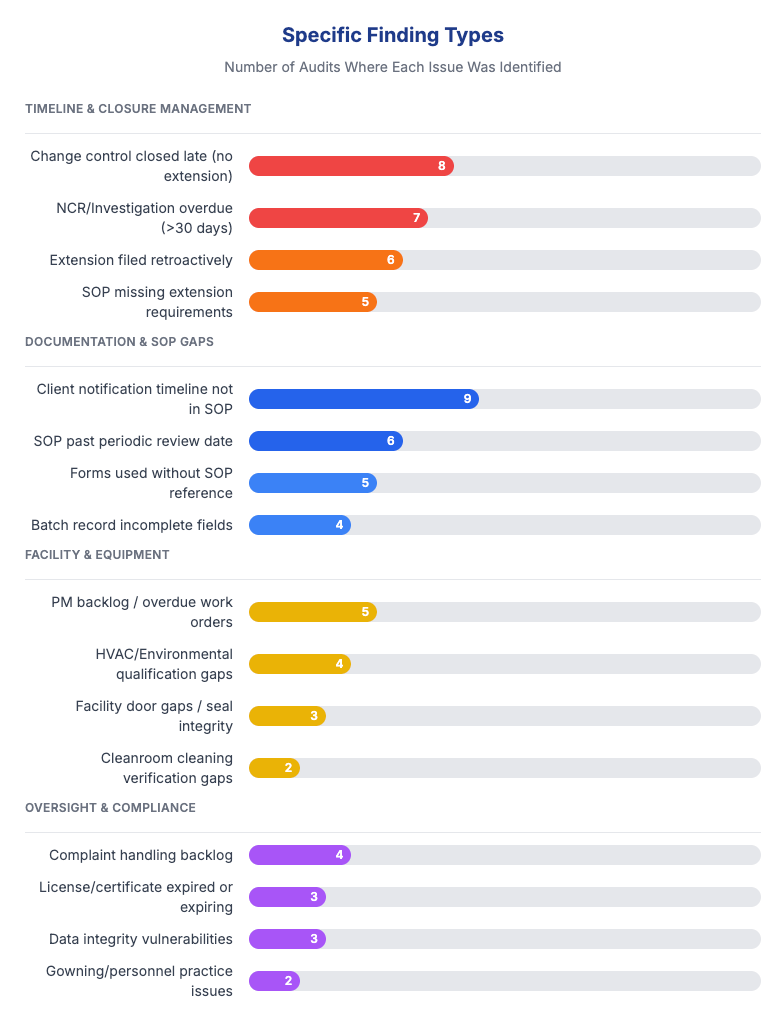
Repeat findings:
20% of audits had findings that recurred from prior years
Recurring issues included: complaint backlogs, facility door gaps, PM backlogs, late QA approvals
Root cause: CAPA effectiveness checks not performed or not defined
Geographic performance:
China: Cleanest outcomes; 3 of 5 audits had zero major findings
India: Consistent; 1–3 minor findings typical; administrative gaps (expired licenses/certificates)
United States: Widest variance; startup facilities showed qualification debt
Ireland: Middle of the pack; procedural alignment gaps between global and local SOPs
Netherlands: Logistics scope only; no significant findings
Patterns to watch:
Supplier notification timelines missing from SOPs — quality agreements require it, procedures don’t address it
Retroactive change control extensions — deadlines missed, extensions approved 30+ days later with no justification
No transport validation — ~30% of facilities rely on logistics providers without product-specific validation
Startup qualification debt — HVAC, EM, media fills pending without consolidated project plans
CAPA effectiveness checks skipped — corrections completed, but no verification they actually worked
What’s working:
Zero critical findings across the entire dataset
Strong environmental monitoring at established CDMOs
Mature document control at ISO-certified facilities
Effective audit trail implementations in validated electronic systems
Open, responsive engagement from audit hosts at closing meetings
Below, we expand on the themes we actually saw in the reports. For each, you'll find: what we observed, how often, representative examples (generalized to protect identity), and prescriptive actions you can take.
1. Documentation and change control remained the biggest gap
In roughly 70% of audits (the same as our prior analysis), we observed some form of documentation gap.
Documentation deficiencies appeared more than any other finding category—not because facilities lacked procedures, but because the procedures weren’t consistently followed or didn’t address common edge cases.
What we found:
Timeline management failures: At multiple sites, investigations and change controls exceeded their procedural timelines without any documented extension request. In one case, a change control initiated in February with a 10-day target closure wasn’t closed until April—nearly two months late—with no extension request ever submitted. The SOP simply didn’t address what to do when a deadline couldn’t be met.
Retroactive extensions that undermined credibility: At another facility, an extension request for an investigation was approved more than 30 days after the original due date had already passed. While the investigation itself was thorough, the retroactive extension approval (with no scientific justification for the delay) would be difficult to defend to an inspector asking, “Why wasn’t this flagged earlier?”
Uncontrolled templates for controlled activities: Management review meetings at one site were documented using PowerPoint presentations that weren’t controlled documents, even though the governing SOP referenced specific templates that should be used. The content of the reviews was appropriate, but the format created a document control gap that could lead to version confusion or incomplete archival.
Missing signatures on critical documents: Business continuity plans and emergency drill reports at one site were complete in content, but didn’t have signatures and dates from the reviewers and approvers who should have formally signed off. This turns a good practice into an undocumented one.
Incorrect or missing data fields: Equipment cleaning records at one site showed “N/A” entered in fields where room numbers were required. The cleaning was performed correctly, but the documentation didn’t demonstrate where the equipment was cleaned—a basic traceability failure.
Forms without attribution: One site’s Deviation Assistance Tool—a form used to document and investigate deviations—did not capture who filled out the document or when. The completed forms were thorough, but without names and dates, the audit trail was incomplete.
We see that FDA investigators routinely pull change control and investigation records and calculate cycle times. Overdue records without extensions suggest a facility that either lacks oversight or tolerates delays.
We see a lot of warning letters specifically cite "failure to thoroughly investigate unexplained discrepancies" and "failure to establish and follow written procedures"—exactly the kind of findings that emerge when documentation discipline breaks down.

A few recommendations:
Build explicit “stop-the-clock” rules into your deviation, OOS, and change control SOPs. Define who may grant an extension, when (before the deadline, not after), what justification is required, and what the maximum allowable extension period is.
Require real-time extension entries with documented scientific or operational rationale and a new committed due date.
Audit your SOP-to-template alignment. Every SOP that references a template should reference a controlled template with a document number, and that template should be easily retrievable.
Implement periodic “documentation hygiene” checks—monthly or quarterly reviews of open records approaching deadlines, with escalation to management for any records past 80% of their target timeframe.
2. Investigations and CAPAs were too slow (or missing entirely)
Observed in roughly 50% of our audits here, even when investigations were eventually completed correctly, the timing often created problems. And in some cases, situations that clearly warranted CAPA investigation never actually triggered one.
What we found:
Chronic overdue investigations: At one site, the NCR (Non-Conformance Report) log showed multiple investigations exceeding the 30-day SOP-mandated closure window, with some remaining open for months. Nine NCR records were found to be over 60 days old with no closure documentation uploaded. A pattern like this usually points to a systemic backlog rather than isolated exceptions.
OOS results not recognized in real time: At one lab we audited, an HPLC-based test for delivered dosage uniformity produced an OOS result, but the investigation wasn’t opened until more than a month later. The analyst had processed the data and moved on without recognizing the OOS. By the time it was discovered during data review, the original test solutions had expired and could no longer be evaluated. This is a good example of a pretty fundamental training gap we encounter quite a bit: analysts must be able to recognize OOS results immediately and escalate them within one business day.
QA oversight failures not treated as CAPA-worthy: At one facility, QA approved an investigation that had been processed on the wrong form. When the error was discovered, a replacement investigation was completed correctly, but no CAPA was opened to address the systemic question: how did QA miss this error in the first place? QA oversight lapses should always trigger their own investigation and corrective action, not just be quietly fixed.
Deviations closed without meaningful CAPAs: At one site, the list of deviations over 12 months filled 3.5 pages, while the list of CAPAs over the same period was only half a page. Simple improvements—”revise the work instruction,” “retrain the operator”—were implemented informally without opening CAPA records, even though the deviation management procedure allowed for it. This pattern suggests CAPA avoidance rather than genuine continuous improvement.
Long-running investigations: One major process incident remained under investigation for more than 18 months after the original event, suggesting resource constraints or unclear ownership.
FDA's OOS guidance (May 2022) emphasizes the importance of timely investigation, specifically noting that delays can compromise the ability to meaningfully investigate.
Warning Letters frequently cite "failure to complete investigation within [X] days as required by your procedure"—a finding that's trivially easy for investigators to document by comparing record dates to SOP requirements.
Make sure you’re tracking investigation and CAPA cycle times as a KPI reported to management review. You should also have clear escalation triggers (e.g., 80% of target timeline) that require management attention before deadlines are missed.
Train analysts explicitly on real-time OOS recognition. Use visual aids in the laboratory showing what an OOS result looks like for common test methods. Require analysts to flag results before leaving for the day.
Define CAPA triggers for QA oversight failures: any error in QA review that reaches downstream processes should automatically generate a CAPA evaluation.
Review your deviation-to-CAPA ratio. A very low ratio may indicate that CAPAs are being avoided rather than appropriately applied. This is a great metric suprisingly few teams keep in their reporting.
3. Facility and equipment qualification gaps kept appearing
Observed in approximately 40% of these audits, these findings were most prominent at sites undergoing expansion, preparing for new product launches, or transitioning between development and commercial phases.
What we found:
Interconnected qualification activities running without coordination: At one facility preparing for commercial manufacturing, the following activities were all in an incomplete or planned state: HVAC qualification, environmental monitoring performance qualification, media fills, facility cleaning procedures, equipment cleaning and decontamination validation, raw material qualification, isolator qualification, temperature and humidity monitoring systems, computer system validation procedures, calibration and maintenance programs, and supplier qualification. While each activity had some level of planning, they weren’t consolidated under a formal Quality Plan with clear owners, dependencies, and deadlines.
HVAC delays cascading through other systems: At the same facility, HVAC qualification was delayed by facility power upgrades, which in turn were delayed by permitting with the local utility. This single dependency held up the environmental monitoring qualification, which in turn prevented baseline establishment for microbiological monitoring during production.
Calibration and PM stickers showing signs of neglect: At multiple sites, calibration stickers were curled, faded, or difficult to read. In one case, a door inspection sticker showed a due date that had passed without the inspection being completed—and without an extension being documented. At another facility, 201 open preventive maintenance (PM) work orders were identified in SAP for the current year, with some PM due dates in the system not aligned with actual performance dates.
Labeling inconsistencies: Storage areas, equipment tags, reagent labels, and shelf designations at some facilities used handwritten labels on colored tape that could smudge or become illegible over time. This creates risk for both traceability (which equipment was cleaned?) and cross-contamination (which storage area is this?).
Equipment without identification: At one medical device facility, a piece of manufacturing equipment lacked the required equipment asset ID tag, even though the SOP required all active equipment to be labeled with identification.
Environmental monitoring without baseline: At one site, previous efforts to establish environmental monitoring baselines had shown passing particulate levels (ISO 7/8) but multiple hits for spore-forming microorganisms. However, there was no baseline data showing passing microbiological results in the current production configuration.
21 CFR 211.42(c) requires that buildings have “suitable size, construction and location to facilitate cleaning, maintenance, and proper operations.”
FDA investigators often look for evidence that equipment is appropriately qualified before use and that environmental conditions are controlled. A site with visible gaps in qualification (incomplete protocols, missing baselines, undated stickers) signals potential control problems throughout the operation.
We suggest most teams consolidate all open qualification activities into a single Quality Plan with clear ownership, deadlines, and dependency mapping. Track this plan in your management reviews.
Define precise verification methods for utilities and equipment calibration, and include those methods in SOPs. Don’t assume operators know how to verify calibration status—write it down!
Implement a labeling standard that requires durable, printed labels with document numbers where appropriate. Eliminate handwritten labeling on equipment and storage areas used for GxP operations.
Establish clear rules for what happens when qualification activities are delayed: who is notified, how is the delay documented, and what interim controls apply.
4. Supplier and vendor oversight was underdeveloped
The gaps here (present in 35% of these audits) weren’t about whether suppliers were qualified, but about whether the procedural infrastructure around supplier oversight was adequate to manage ongoing relationships and change notifications.
What we found:
Client notification timelines not specified in SOPs: At multiple CDMO sites, SOPs for supplier change notification did not clearly define the timeframes for notifying clients. In one case, the quality agreement with the client required notification of supplier changes within 5 business days, but the internal SOP was silent on timing. This mismatch creates risk: the CDMO may comply with its own SOP while violating the quality agreement.
Stage-specific requirements missing: One SOP addressed notification requirements for PPQ and commercial projects, but didn’t clearly apply to clients with products still in clinical development. A change affecting a Phase 2 clinical product might not be subject to the same notification rigor as a commercial product, even though the clinical sponsor needs the information.
Quality agreements without regulatory specificity: Some quality agreements we reviewed didn’t clearly specify which regulatory standards (GMP, GDP, IPEC, etc.) applied to the supplier relationship. This creates ambiguity about what “compliance” means under the agreement.
Major changes not communicated: At one API manufacturing site, a major change—incorporating nitrosamine impurity testing into the API specification—was not communicated to the customer as required by the quality agreement. At another site, changes related to processes, test specifications, and product specifications were implemented without customer notification, even though the quality agreement explicitly required notification of “major changes.”
